臨床試験は健康な人・患者さんを対象に3段階に分けて行なわれます

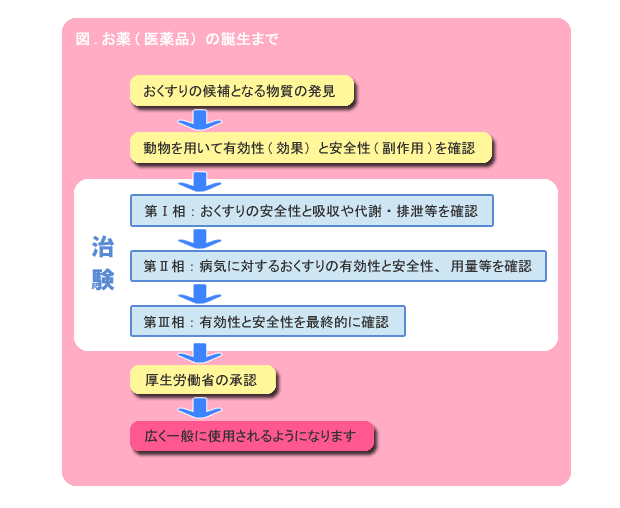
新しい薬を開発するためには、動物を用いた前臨床試験で薬の候補物質の薬効と安全性を十分に確認したうえで、人体での主作用と副作用を検証する必要があります。そのために実施される臨床試験が「治験」です。
治験は厚生労働省が定めた基準、すなわちGCP(医薬品臨床試験の実施基準)に従って行なわれます。具体的には第Ⅰ相から第Ⅲ相までの臨床試験を順番に実施し、得られた結果を整理して承認申請資料を作成し、厚生労働省に提出します。
第Ⅰ相試験はフェーズ・ワンとも呼ばれ、少数の健康な成人を対象に薬剤を投与し、薬効よりも安全性や薬物の体内動態(体内に吸収された薬物が血流を解して各組織に分布され、効果を示し、体外に排出されるまでの一連の動きのこと)を確認します。
第Ⅱ相試験では、同意を得た患者さんに投与し、効果のある病気や病態を調べるとともに、使い方(投与量・投与方法など)の違いによる効果の比較も行なわれます。
最終段階の第Ⅲ相は製造承認を得るための試験で、できるだけ多くの患者さんを被験者として、薬剤の有効性、安全性、使い方の再確認が実施されます。
治験は2つのグループに分けて行なわれます。実際に検討する薬剤と、何の効果もないプラセボ(偽薬)をそれぞれのグループに同じ用量、同じ条件で投与し、その経過と結果を評価します。ただし、グループ分けされていることは被験者には知らされません。これを二重盲検法といいます。
また、被験者の無作為選択、つまり選択条件や枠組みを設定せず、自然に任せて集めなければなりません。こうした公平な治験環境を設定することを「バイアス(偏り)を除く」と表現します。
第Ⅰ〜Ⅲ相までの治験が無地に終了し、薬の候補物質の有効性と安全性が証明されたならば、いよいよ厚生労働省に対して承認申請を行うことになります。
新薬の承認審査では、厚生労働省の外郭団体である独立行政法人・医薬品医療機器総合機構が臨床試験実施基準の適合性調査や。データ整合性の確認を行い、続いて厚生労働省の薬事・食品衛生審議会医薬品第二部会が、商人を与えるかどうかの判断を行います。
この作業は膨大であり、かつ慎重に進められるため、審査プロセスには従来2年前後の時間を要していました。しかし、最近の審査迅速化を望む世論に押される形で、現在では1年程度に短縮化が測られています。
治験に参加する患者の権利と安全を確保するインフォームドコンセント

治験におけるインフォームド・コンセントとは、治験の目的や薬の効果・副作用、その投与方法、プライバシーなどが記載された同意説明文書をもとに、担当医師がわかりやすい言葉で説明し、患者さんがその内容をよく理解したうえで、「自由意志」により治験に参加することに同意するということです。
例えば、患者さんが薬のことはよくわからないからといって、「先生に全てお任せします」といって、治験の内容をよく理解せずに署名したり、逆に医師があいまいな説明で患者さんを説得して同意させた場合は、インフォームド・コンセントとは言いません。
患者さんは、疑問があれば質問し、納得いくまで説明を受けることができます。その場で治験への参加を決める必要はなく、家族に相談して判断をすることもできます。治験への参加に同意しないことも患者さんの権利ですので、無理にすすめられることはありません。
参加の同意説明文書へのサインはあくまでも患者さんの自由意志によるものであって契約ではありませんので、サインをした後に途中で止めたくなった場合は、その理由の如何にかかわらず、いつでも治験参加を取りやめることができます。
参加を取りやめても患者さんへの不利益は一切ない旨が法令で定められています。
文書によるインフォームド・コンセントは、医師と患者さんが対等な立場で話し合い、治験に参加する患者さんの権利と安全性を確保する意味で欠かせない手続きとなっています。